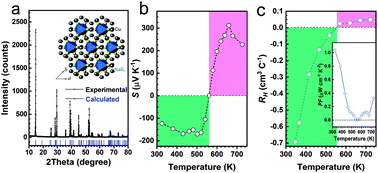
J. Alloys Compd., 2020, 821, 153482. https://doi.org/10.1016/j.jallcom.2019.153482
Ca-α-Sialon: Eu2+, a well-known yellow phosphor, has been widely studied due to its broad UV-blue excitation with high quantum efficiency. Herein, we report the yellow persistent luminescence (PersL) of a series of Ca-α-Sialon: Eu2+ compounds with chemical formula CaSi10-nAl2+nOnN16-n: xEu2+ (m = 2, n = 0∼1, x = 0.1%∼8%) prepared by high-temperature solid-state method. Upon 254 nm ultraviolet excitation, Ca-α-Sialon: Eu2+ shows yellow PersL, and the persistent time is strongly dependent on the Eu2+ and oxygen concentrations. The best persistent time is measured to be about 60 min for the CaSi10Al2N16: 0.5% Eu2+ sample. A very broad trap depth distribution, i.e. 0.6–1.4 eV, originating from two categories, are obtained by analyzing preheating thermoluminescence (TL) spectra using initial rise method. Comparing thermoluminescence excitation spectra (TLEs) with photoluminescence excitation spectra (PLEs), we verify that electrons as charge carriers are excited from 4f ground state of Eu2+ to conduction band (CB) directly in the charging process for PersL. By functional theory (DFT) calculations, we verify that the trap levels responsible for PersL are impurity defects including VO and VN in Ca-α-Sialon. Furthermore, the PersL mechanism is given on the basis of constructing the host referred binding energy (HRBE) diagram. By virtue of NIR photo-stimulating PersL spectrum, we demonstrate that Ca-α-Sialon has a potential application in anti-counterfeit and information storage. This work would encourage more exploration of Eu2+-doped nitride phosphor for persistent or long-persistent luminescence.